- Home
- >
- Articles
Dermatology Articles
Acne Treatments

Severe acne is difficult to treat, but may be manageable. The treatment will focus on preventing future acne outbursts. There are various types of acne treatments; some target the surface of the skin, while others will be systemic. The type of treatment will be established according to the severity ... Continue Reading
Severe acne is difficult to treat, but may be manageable. The treatment will focus on preventing future acne outbursts. There are various types of acne treatments; some target the surface of the skin, while others will be systemic. The type of treatment will be established accord .. Continue Reading

Everyone has unique skin, which is why there are so many different types of acne treatment solutions on the market and in the home remedy sections of books. If you haven’t tried using the healing, curing and preventing effects of strawberries before, now is the time. For many people, these homemad ... Continue Reading
Everyone has unique skin, which is why there are so many different types of acne treatment solutions on the market and in the home remedy sections of books. If you haven’t tried using the healing, curing and preventing effects of strawberries before, now is the time. For many p .. Continue Reading
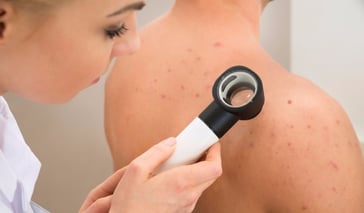
Acne can be an unsightly, but a harmless problem. Acne is due to a problem in the pilosebaceous unit, which is found all over the body, with some exceptions such as the soles of the feet, palms and a few others. There are a few ways to cure acne, which usually involve antibiotic medication or natura ... Continue Reading
Acne can be an unsightly, but a harmless problem. Acne is due to a problem in the pilosebaceous unit, which is found all over the body, with some exceptions such as the soles of the feet, palms and a few others. There are a few ways to cure acne, which usually involve antibiotic .. Continue Reading

Out of the many acne treatment options available, Ayurvedic and home remedies make use of sandalwood. Sandalwood is an evergreen medium-sized tree, which has mature bark with a pleasant fragrance. Like turmeric powder, this is also used in most of the beauty or skin care products. In oily skin, dirt Continue Reading
Out of the many acne treatment options available, Ayurvedic and home remedies make use of sandalwood. Sandalwood is an evergreen medium-sized tree, which has mature bark with a pleasant fragrance. Like turmeric powder, this is also used in most of the beauty or skin care products .. Continue Reading
Scar Removal
Scarring is the natural result of injury to the skin. Whenever an injury affects the deeper layers of the skin, the body produces collagen fibers to heal the damaged area. The new tissue that is formed is known as a scar. This new tissue is different than normal skin; it is less resistant to UV dama ... Continue Reading
Scarring is the natural result of injury to the skin. Whenever an injury affects the deeper layers of the skin, the body produces collagen fibers to heal the damaged area. The new tissue that is formed is known as a scar. This new tissue is different than normal skin; it is less .. Continue Reading

When you finally decide to get a tattoo, it is important that you choose a reputable tattoo artist. Tattoo scarring is caused by a possible handful of reasons, and one of those reasons is having a bad tattooist. However, it is also important to know that a tattoo is a wound and wounds leave scars; h ... Continue Reading
When you finally decide to get a tattoo, it is important that you choose a reputable tattoo artist. Tattoo scarring is caused by a possible handful of reasons, and one of those reasons is having a bad tattooist. However, it is also important to know that a tattoo is a wound and w .. Continue Reading

To prevent scarring, there are a wide range of traditional and alternative remedies. Aloe vera is among the most frequently mentioned solutions used on the skin to reduce swelling, scars or prevent scarring. Aloe Vera Benefits Aloe vera is a juicy plant that typically thrives in arid climates and is Continue Reading
To prevent scarring, there are a wide range of traditional and alternative remedies. Aloe vera is among the most frequently mentioned solutions used on the skin to reduce swelling, scars or prevent scarring. Aloe Vera Benefits Aloe vera is a juicy plant that typically thrives in .. Continue Reading
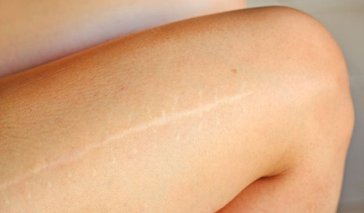
Scarring can occur when a mark is left after the healing of a wound or after an injury to the skin’s surface, maybe by a burn or sometimes from a medical procedure. A scar is a biological repair of the body’s tissues and skin, and is a natural healing process. There are typically three types of ... Continue Reading
Scarring can occur when a mark is left after the healing of a wound or after an injury to the skin’s surface, maybe by a burn or sometimes from a medical procedure. A scar is a biological repair of the body’s tissues and skin, and is a natural healing process. There are typic .. Continue Reading
Anti-Aging Treatments

Although you have no control over the genetic rate that you will age, there are things you can do to prevent fine lines and wrinkles from developing too soon. Exercise and Maintain a Balanced Diet Exercising regularly and eating a balanced diet are important in maintaining a healthy vitality and a y ... Continue Reading
Although you have no control over the genetic rate that you will age, there are things you can do to prevent fine lines and wrinkles from developing too soon. Exercise and Maintain a Balanced Diet Exercising regularly and eating a balanced diet are important in maintaining a heal .. Continue Reading

Aging is a natural part of life. Many people try to fight aging’s effect on their skin and general facial appearance with beauty products, scrubs and facials, various dermal injections and plastic surgery. Before jumping into a quick fix solution I recommend my patients develop a thorough understa ... Continue Reading
Aging is a natural part of life. Many people try to fight aging’s effect on their skin and general facial appearance with beauty products, scrubs and facials, various dermal injections and plastic surgery. Before jumping into a quick fix solution I recommend my patients develop .. Continue Reading

There are a variety of skin care products out there that insist they have the cure for wrinkles. The truth is there is no way to prevent fines lines and wrinkles from developing as they are a part of the natural part of aging process. But, there are things we can do to help slow down the process. He ... Continue Reading
There are a variety of skin care products out there that insist they have the cure for wrinkles. The truth is there is no way to prevent fines lines and wrinkles from developing as they are a part of the natural part of aging process. But, there are things we can do to help slow .. Continue Reading

Liquid facelifts reduce signs of aging and restore the facial contours of youth to look rested and relaxed. Learn how Liquid facelifts can help you. Continue Reading
Liquid facelifts reduce signs of aging and restore the facial contours of youth to look rested and relaxed. Learn how Liquid facelifts can help you. Continue Reading
Skin Conditions

A cold sore is a small red blister that develops on the upper or lower lip. Before the cold sore occurs, you will feel a tingle on the lip. The cold sore may be painful and you may also notice that it may burst or drain. When the cold sore starts to heal, it may be itchy. Cold sores may occur if you Continue Reading
A cold sore is a small red blister that develops on the upper or lower lip. Before the cold sore occurs, you will feel a tingle on the lip. The cold sore may be painful and you may also notice that it may burst or drain. When the cold sore starts to heal, it may be itchy. Cold so .. Continue Reading

There is no known cure for eczema, but there are a variety of treatment options available to help alleviate the symptoms of eczema and reduce the frequency of flare-ups. The following is a list of the most common treatment options for eczema: Topical Corticosteroids This type of topical medication i ... Continue Reading
There is no known cure for eczema, but there are a variety of treatment options available to help alleviate the symptoms of eczema and reduce the frequency of flare-ups. The following is a list of the most common treatment options for eczema: Topical Corticosteroids This type of .. Continue Reading

Chapped lips, or chronic chapped lips (known as Cheilitis), is the condition when the lips become dry or cracked. Most often this occurs because of outside stimulus, such as cold or dry weather, licking your lips, dehydration or an allergic reaction. Healing Chapped Lips with Hydrocortisone Cream Tr ... Continue Reading
Chapped lips, or chronic chapped lips (known as Cheilitis), is the condition when the lips become dry or cracked. Most often this occurs because of outside stimulus, such as cold or dry weather, licking your lips, dehydration or an allergic reaction. Healing Chapped Lips with Hyd .. Continue Reading

Psoriasis may have several symptoms depending on the type of psoriasis. The most common symptom is red patches on the skin covered with dead skin cells. While there is no cure for psoriasis, certain treatments may help to reduce symptoms and improve quality of life. Topical Medications Certain topic ... Continue Reading
Psoriasis may have several symptoms depending on the type of psoriasis. The most common symptom is red patches on the skin covered with dead skin cells. While there is no cure for psoriasis, certain treatments may help to reduce symptoms and improve quality of life. Topical Medic .. Continue Reading
Laser & Light Treatments

Active FX heals sun damaged skin. The Active FX laser fixes fine lines, wrinkles, freckling, and other signs of aging. Find an Active FX doctor near you. Continue Reading
Active FX heals sun damaged skin. The Active FX laser fixes fine lines, wrinkles, freckling, and other signs of aging. Find an Active FX doctor near you. Continue Reading

If you're considering laser tattoo removal and you're currently pregnant or hope to soon become pregnant, it's important to consider whether this procedure is safe for you and your child. Be sure to disclose your pregnancy status to your cosmetic specialist. During Pregnancy You should not have lase ... Continue Reading
If you're considering laser tattoo removal and you're currently pregnant or hope to soon become pregnant, it's important to consider whether this procedure is safe for you and your child. Be sure to disclose your pregnancy status to your cosmetic specialist. During Pregnancy You .. Continue Reading
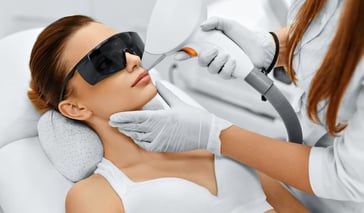
Laser energy treatments remove unwanted hair and slow the growth of new hair. Learn if you're a good candidate and find a laser hair removal specialist. Continue Reading
Laser energy treatments remove unwanted hair and slow the growth of new hair. Learn if you're a good candidate and find a laser hair removal specialist. Continue Reading
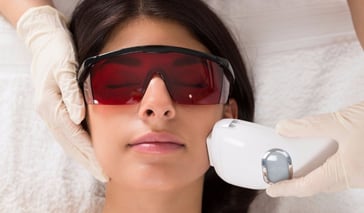
Tighten sagging skin and reduce the signs of wrinkles and aging with the Skin Tyte laser treatment. Learn how a Skin Tyte specialist can help you. Continue Reading
Tighten sagging skin and reduce the signs of wrinkles and aging with the Skin Tyte laser treatment. Learn how a Skin Tyte specialist can help you. Continue Reading
Cellulite Treatments

Cellulite is a common problem affecting mostly women, but may be met in men also. Even though cellulite does not cause pain, it is considered unaesthetic and women dread the thought of having to deal with this problem. In some cases, cellulite can be prevented. However, in order to prevent cellulite ... Continue Reading
Cellulite is a common problem affecting mostly women, but may be met in men also. Even though cellulite does not cause pain, it is considered unaesthetic and women dread the thought of having to deal with this problem. In some cases, cellulite can be prevented. However, in order .. Continue Reading

Does cellulite cream really work? There are many cellulite creams available for consumers to purchase that work. To locate these creams, it may require you to conduct a small amount of research regarding these products. You must also remember that when you are using body enhancing products, such as ... Continue Reading
Does cellulite cream really work? There are many cellulite creams available for consumers to purchase that work. To locate these creams, it may require you to conduct a small amount of research regarding these products. You must also remember that when you are using body enhancin .. Continue Reading

Cellulite is a very common skin condition recognized by a dimpled, bumpy appearance on the skin. Cellulite occurs when fat cells become too large for their tissue compartments, causing the skin to bulge. There are a variety of factors that can contribute to causing cellulite. These include: Hormones Continue Reading
Cellulite is a very common skin condition recognized by a dimpled, bumpy appearance on the skin. Cellulite occurs when fat cells become too large for their tissue compartments, causing the skin to bulge. There are a variety of factors that can contribute to causing cellulite. The .. Continue Reading

Cellulite is an issue for virtually every woman at some point or another over the course of her life. Cellulite tends to develop in the thighs, upper legs and buttocks, and causes unsightly changes to the way that the skin appears. Many women spend hundreds of dollars on expensive creams and ointmen ... Continue Reading
Cellulite is an issue for virtually every woman at some point or another over the course of her life. Cellulite tends to develop in the thighs, upper legs and buttocks, and causes unsightly changes to the way that the skin appears. Many women spend hundreds of dollars on expensiv .. Continue Reading
All Article Categories
Suggested Doctors
Recently Asked Questions
Affiliations
